Importancia de la RM fetal en el diagnóstico precoz de esclerosis tuberosa.
Palabras clave:
RM, RM fetal, esclerosis, tuberosa, diagnóstico, precoz, poster, seramResumen
-
Analizar los hallazgos ecográficos prenatales en el estudio de la esclerosis tuberosa y describir sus limitaciones técnicas.
-
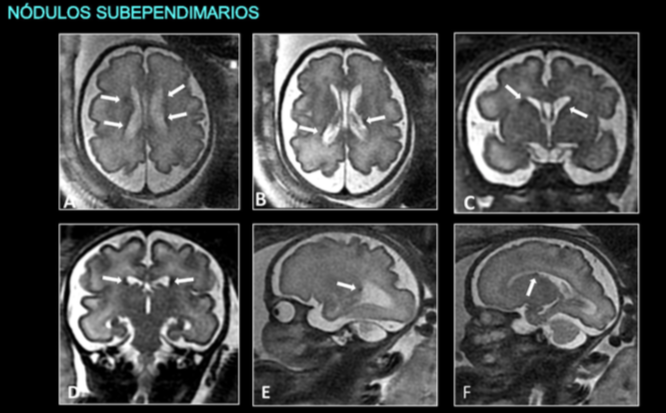
Explicar los hallazgos radiológicos característicos de la esclerosis tuberosa en RM fetal, haciendo hincapé en la importancia del diagnóstico prenatal por RM ante un feto con rabdomioma cardíaco.
-
Valorar el papel de la RM postnatal para confirmar los hallazgos radiológicos prenatales y establecer el grado de afectación cerebral.
Revisión del tema
ESCLEROSIS TUBEROSA
La esclerosis tuberosa (ET) o enfermedad de Bourneville es un síndrome neurocutáneo que que afecta a varios órganos como el cerebro, la piel, los riñones, los ojos y el corazón, y se caracteriza por presentar múltiples tumores benignos (hamartomas o tumores de bajo grado). Es la segunda facomatosis más común después de la neurofibromatosis tipo1.
Incidencia
La ET tiene una herencia autosómica dominante con expresión variable y penetrancia incompleta. Su incidencia es de 1 cada 10000 y su prevalencia 1 por 6000 a 10000 nacidos vivos. Afecta por igual a todas las razas y a ambos sexos.
Etiología
Se han identificado mutaciones en dos genes diferentes: TSC1, en el cromosoma 9q34, y TSC2, en el cromosoma16p13. Las pruebas genéticas para los genes TSC1 y TSC2 genes del líquido amniótico o del muestreo de vellosidades coriónicas pueden ser definitivas en el 80% de las mutaciones. Sin embargo,
debido a la heterogeneidad genética, las pruebas pueden tener una tasa de falsos negativos del 10% al 20%.
El TSC1 codifica una proteína llamada hamartina, cuya principal función parece ser la de formar complejos con la proteína tuberina, codificada por el gen TSC2. Este complejo funciona como un supresor tumoral mediante la retroalimentación negativa de la cascada de la mTOR quinasa, cuya actividad es importante en la regulación del crecimiento y la proliferación celular. El aumento de la activación de mTOR desencadena un proliferación celular desorganizada con diferenciación celular anómala. Existe una alta penetrancia de la anomalía genética, pero la expresión fenotipo es variable. Aproximadamente 70% a 80% de los casos son esporádicos, como resultado de mutaciones de novo.
Las mutacionesTSC2 son aproximadamente cinco veces más comunes que las mutaciones de TSC1. Las mutacionesTSC1 son un poco menos comunes en los casos esporádicos y son algo más comunes(15% -50%) en los casos familiares. El gen TSC2 se encuentra sólo en 48 pares de bases del ADN del gen de la enfermedad renal poliquística del adulto (PKD1). Cuando los pacientes tienen una deleción del gen TSC2 y PKD1, tienen un fenotipo particularmente grave con un inicio muy precoz de PKD1 y de insuficiencia renal. Los individuos con mutaciones de TSC1 parecen tener menos riesgo de retraso mental y menor frecuencia de convulsiones. Presentan menor número de nódulos subependimarios y tubers corticales, menos hamartomas retinianos, una enfermedad renal leve, y menores angiofibromas faciales. Aquellos con mutaciones de TSC2 probablemente tienen un mayor riesgo de desarrollar quistes renales. Sin embargo, algunas mutaciones TSC2 tienen fenotipos relativamente suaves, probablemente debido a que el efecto de la mutación en la función de la tuberina es el último factor en la determinación de la gravedad del fenotipo.
Antes de concluir que un niño afectado presenta una nueva mutación, es necesario examinar ambos padres a fondo, incluyendo el examen cutáneo con lámpara de Wood o de luz ultravioleta , un estudio de neuroimagen y, si es posible, el análisis cromosómico de múltiples tejidos, incluyendo las células germinales. Algunos individuos adquieren el ET a través de un proceso llamado mosaicismo gonadal. Esto significa que los padres de estos pacientes no presentan defectos evidentes en los dos genes que causan el trastorno. Sin embargo, los padres pueden tener un niño que padezca ET porque una parte de una de las células reproductoras de uno de los padres (el esperma o los óvulos) puede contener la mutación genética sin que las otras células del cuerpo estén involucradas. En casos de mosaicismo gonadal, la prueba genética de una muestra de la sangre podría no revelar el potencial de transmisión de la enfermedad a los descendientes.
El riesgo de recurrencia en futuros embarazos es del 2% al 3% si no se demuestra afectación de los padres y del 50% si uno de los progenitores padece ET.
Descargas
Citas
Goel R, Aggarwal N, Lemmon ME, Bosemani T. Fetal and maternal manifestations of tuberous sclerosis complex: Value of fetal MRI. Neuroradiol J. 2016 Feb 2. pii: 1971400915621323.
Jurko T, Jr AJ, Krsiakova J, Jurko A, Minarik M, Matasova K, Zibolen M. Prenatal diagnosis of cardiac rhabdomyoma associated with tuberous sclerosis: report of 3 cases. Neuro Endocrinol Lett. 2015 Dec;36(6):521-3.
Paleologou E, Nicholl R. Uncommon antenatal presentation of tuberous sclerosis. BMJ Case Rep. 2014 Jun 23;2014.
Blondiaux E1, Garel C. Fetal cerebral imaging - ultrasound vs. MRI: an update. Acta Radiol. 2013 Nov;54(9):1046-54.
Staley BA, Vail EA, Thiele EA.Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011 Jan;127(1):e117-25.
Isaacs H. Perinatal (fetal and neonatal) tuberous sclerosis: a review. Am J Perinatol. 2009 Nov;26(10):755-60.
Martín Fernández-Mayoralas D, Fernández-Jaén A, Muñoz-Jareño N, Calleja-Pérez B, Villó-Siresol N. El complejo de esclerosis tuberosa: importancia del examen de la piel en la epilepsia. Acta Pediatr Esp. 2009; 67: 231-233.
Wortmann SB, Reimer A, Creemers JW, Mullaart RA. Prenatal diagnosis of cerebral lesions in tuberous sclerosis complex (TSC). Case report and review of the literature. Eur J Paediatr Neurol. 2008; 12: 123-126.
Prats-Viñas JM. Facomatosis que cursan con manchas acrómicas, esclerosis tuberosa de Bourneville. Criterios diagnósticos y protocolo de
seguimiento. Rev Neurol. 1996; 24: 1.056-1.059.
Janeiro PC, Cunha MS, Cordeiro I, Santos HG, Antunes NL. Ocurrencia simultánea de neurofibromatosis y esclerosis tuberosa, adquiridas como neomutaciones. Rev Neurol. 2008; 46: 347-350.
Tuchman RF, Moshe SL, Rapin I. Trastornos del neurodesarrollo y epilepsia. Rev Neurol. 2005; 40 Supl 1: 3-10.
Pascual-Castroviejo I. Neurosurgical treatment of tuberous sclerosis complex lesions. Childs Nerv Syst. 2011; 27: 1.211-1.219.
Muhler MR, Rake A, Schwabe M, Schmidt S, Kivelitz D, Chaoui R, et al. Value of fetal cerebral MRI in sonographically proven cardiac rhabdomyoma. Pediatr Radiol. 2007; 37: 467-474.
Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004; 19: 643-649.
Artigas-Pallares J, Gabau-Vila E, Guitart-Feliubadalo M. El autismo sindrómico (II). Síndromes de base genética asociados a autismo. Rev Neurol. 2005; 40 Supl 1: 151-162.